Research Interests
Overview
Vision is the most important of our senses. It enables us to richly experience and interact with the world around us. Inherited diseases of the visual system that lead to partial or total blindness severely diminish the quality of life and present a serious social and economic burden.
Research in my laboratory is directed toward identifying and characterizing vertebrate retinal photoreceptor proteins and elucidating their role in 1) phototransduction and related signal transduction pathways; 2) photoreceptor cell structure and morphogenesis; 3) retinal cell-cell and cell-extracellular matrix interactions; and 4) various inherited retinal degenerative diseases including retinitis pigmentosa and macular degeneration that are the leading causes of blindness in the developed world. We are also developing and applying new technology to the study of membrane proteins and treatments for retinal degenerative diseases. To accomplish these goals, we are utilizing a variety of current and emerging biochemical, biophysical, immunochemical, molecular and cell biology approaches to 1) purify and reconstitute membrane proteins into lipid vesicles for structure-function analyses; 2) identify and characterize important structural, functional, and regulatory protein domains; 3) define specific protein-protein interactions responsible for the formation of macromolecular assemblies; 4) develop novel reagents and cell and animal models for analysis of retinal degenerative diseases; and 5) evaluate diagnostic and therapeutic interventions for selective retinal degenerative diseases.
Some of the techniques currently being employed in my laboratory include:
- Generation and characterization of monoclonal antibodies
- cDNA cloning, subcloning and sequencing
- Heterologous protein expression in bacteria, yeast and mammalian cells
- Construction of mutant and chimeric proteins using PCR
- Yeast two hybrid analysis
- Spectrophotometric analysis
- Immunoaffinity and conventional column chromatographyp
- HPLC peptide and FPLC protein chromatography
- Proteomic analysis using mass spectroscopy
- Peptide Synthesis
- Immunoprecipitation and immunoaffinity chromatography
- SDS gel electrophoresis and Western blotting
- Immunocytochemical labeling for fluorescent light microscopy
- Confocal scanning laser microscopy
- Transmission and scanning electron microscopy
- Enzyme and transport assays
- Computer data analysis
- Cell and subcellular fractionation
- Cell and tissue culture techniques
- Cell and tissue culture techniques
- RNA extraction and Northern blotting
- Analysis of transgenic and knockout animals
- Gene therapy
- Drug discovery
The experimental systems we work on and current research areas of interest are briefly described below.
Rod and Cone Photoreceptor Cells
Rod and cone photoreceptor cells of the vertebrate retina are specialized neurons that function in the primary events of the vision. Rod photoreceptors are highly sensitive to light and operate under dim lighting conditions, while cone photoreceptors function under normal light intensity and are responsible for color vision and high visual acuity. Both cells are similar in structure consisting of an outer segment, inner segment, cell body and synaptic region (Fig 1).

The outer segment of rod and cone photoreceptor cells is a specialized subcellular compartment where light is captured and converted into electrical signals in the process known as phototransduction. This unique compartment consists of a stack of 500-2000 highly ordered discs surrounded by a plasma membrane. Each disc is comprised of two closely spaced flattened membranes that are fully (rods) or partially (cones) circumscribed by a hairpin membrane region called the disc rim (Fig 2). 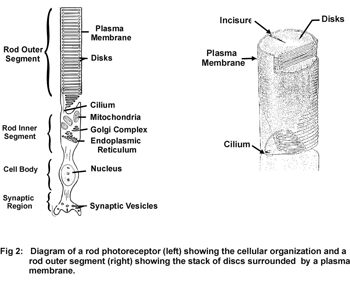
In one area of study, we are identifying proteins and molecular and cellular mechanisms that are responsible for the formation and structural stability of the outer segments. Peripherin-2 and rom-1 are two homologous membrane proteins that play key roles in disc morphogenesis (Fig 3).
These proteins assemble into large disulfide-linked multimeric complexes that are crucial for forming the hairpin rim region of rod and cone outer segment discs. (Fig 3).
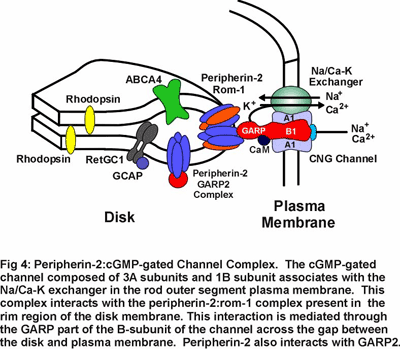
Mutations in peripherin-2 that cause protein misfolding and defective subunit assembly result in abnormal outer segment disc morphogenesis and photoreceptor degeneration. Over 40 different mutations in peripherin-2 are known to cause autosomal dominant retinitis pigmentosa and macular degeneration (see Fig 8). We are continuing to examine the 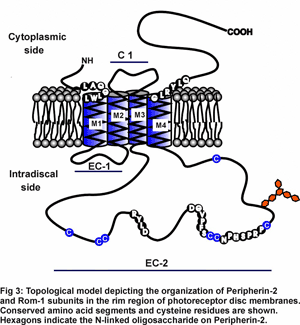 molecular properties and structure of peripherin-2:rom-1 complexes and identifying protein-protein interactions in an effort to define the role of these proteins in the formation and stabilization of the outer segment and mechanisms underlying diseases linked to peripherin-2. Our recent studies indicate that the peripherin-2:rom-1 complex in the rim region of the disc interacts with the cGMP-gated channel:Na/Ca-K exchanger complex in the plasma membrane to form a large molecular assembly of proteins (Fig 4). The cGMP-gated channel is a multi-subunit protein consisting of 3A subunits and 1 B subunit. We are studying the molecular properties of the channel:exchanger complex and its interaction with the peripherin-2:rom-1 complex using a variety of biochemical and molecular biology techniques to define the role of this protein assembly in outer segment structure and function.
molecular properties and structure of peripherin-2:rom-1 complexes and identifying protein-protein interactions in an effort to define the role of these proteins in the formation and stabilization of the outer segment and mechanisms underlying diseases linked to peripherin-2. Our recent studies indicate that the peripherin-2:rom-1 complex in the rim region of the disc interacts with the cGMP-gated channel:Na/Ca-K exchanger complex in the plasma membrane to form a large molecular assembly of proteins (Fig 4). The cGMP-gated channel is a multi-subunit protein consisting of 3A subunits and 1 B subunit. We are studying the molecular properties of the channel:exchanger complex and its interaction with the peripherin-2:rom-1 complex using a variety of biochemical and molecular biology techniques to define the role of this protein assembly in outer segment structure and function.
Phototransduction
Phototransduction in rod outer segments is initiated when the photoreceptor protein rhodopsin captures a photon of light converting the 11-cis retinal chromophore to its all-trans isomer (Fig 5). This reaction leads to a conformational change in rhodopsin and activation of the G-protein coupled visual cascade on the surface of disc membranes. The activation of phosphodiesterase and the resulting decrease in cGMP leads to the closure of cGMP-gated channels in the plasma membrane of rod outer segments to the influx of Na+ and Ca2+ ions and a hyperpolarization of the rod cell. The cell is subsequently returned to its dark state through a series of biochemical reactions that inactivate rhodopsin and the visual cascade system and resynthesize cGMP. The increase in cGMP levels results in a reopening of cGMP-gated channels and a return of the photoreceptor cell to its dark, depolarized state. (Fig 4).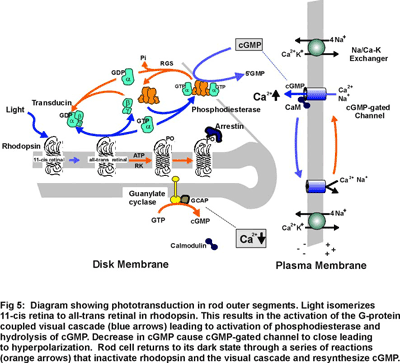
Research in my laboratory is directed toward determining the molecular structure, function and regulation of several key components of the phototransduction process. Current emphasis is directed toward identifying domains in the cGMP-gated channel and Na/Ca-K exchanger that contribute to their structure, function and regulation. We are also examining protein-protein interactions involving glutamic acid rich proteins called GARPs that may contribute to phototransduction and/or light adaptation or help organize the cGMP-gated channel complex within the rod outer segment plasma membrane (see Fig 4). In addition, we are investigating proteins that may be involved in the recycling of all-trans retinal to its 11 cis isomer as part of the regeneration of rhodopsin. As part of these studies, we are carrying a proteomic study of rod and cone outer segments to identify novel proteins which contribute to outer segment structure, function and morphogenesis.
Retinal Degenerative Diseases
Retinitis pigmentosa (RP) and macular degeneration (MD) are a heterogeneous group of inherited retinal degenerative diseases that represent a major cause of blindness in the developed world. RP is characterized by the progressive loss in peripheral and night vision followed by the gradual loss in central vision,  often leading to complete blindness. It is typically associated with the early degeneration of rod photoreceptor cells and subsequent loss in cone photoreceptors. MD is characterized by the marked decrease in central vision and visual acuity with some preservation of peripheral vision. It is generally associated with the degeneration of central cone and rod photoreceptor cells and underlying retinal pigment epithelial cells with preservation of peripheral rods and cones. To date mutations in a large number of genes encoding proteins expressed in rod and cone photoreceptor cells have been linked to various forms of RP and juvenile MD. These include rhodopsin, phosphodiesterase, guanylate cyclase, peripherin-2, ABCR, cGMP-gated channel, RS1 or retinoschisin, and others (Fig 6).
often leading to complete blindness. It is typically associated with the early degeneration of rod photoreceptor cells and subsequent loss in cone photoreceptors. MD is characterized by the marked decrease in central vision and visual acuity with some preservation of peripheral vision. It is generally associated with the degeneration of central cone and rod photoreceptor cells and underlying retinal pigment epithelial cells with preservation of peripheral rods and cones. To date mutations in a large number of genes encoding proteins expressed in rod and cone photoreceptor cells have been linked to various forms of RP and juvenile MD. These include rhodopsin, phosphodiesterase, guanylate cyclase, peripherin-2, ABCR, cGMP-gated channel, RS1 or retinoschisin, and others (Fig 6).
A major effort in my laboratory is being directed toward understanding how disease causing mutations in photoreceptor specific proteins affect their structure, function, expression and subcellular targeting and how defective proteins lead to rod and/or cone photoreceptor cell death. We are now directing our efforts toward characterizing the structure and function of wild-type and mutant forms of several proteins linked to degenerative diseases. ABCA4 (ABCR), a photoreceptor-specific ATP binding cassette (ABC) transporter that has been linked to Stargardt macular dystrophy, cone rod dystrophy and age-related macular dystrophy. The function of ABCA4 is not known although it has been suggested to transport or flip of N-retinylidene-phosphotidylethanolamine across photoreceptor disc membranes (Fig 7)
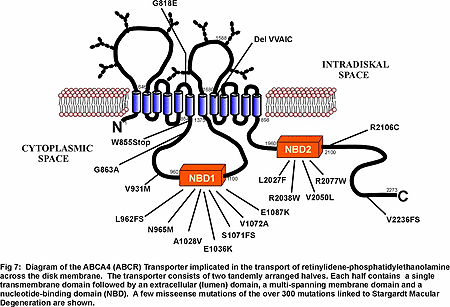
We are now developing methods to identify the substrate transported by ABCA4 and elucidate the mechanism of transport. Peripherin-2 is a member of the family of tetraspanning membrane proteins and has been linked to various forms of autosomal dominant retinitis pigmentosa and macular dystrophy (Fig 8). 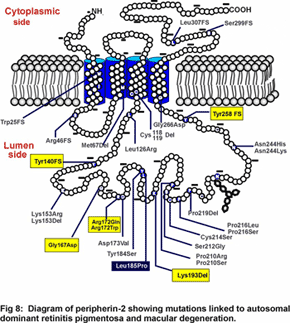 We are investigating the role of this protein in outer segment structure and morphogenesis. RS1 or retinoschisin is a discoidin containing extracellular protein implicated in retinal cell adhesion and associated with X-linked juvenile retinoschisis (Fig 9). We are studying the structure of this protein and how mutations affect its folding pattern and subunit assembly. As part of our research program, we are also carrying out genomic and proteomic studies to identify genes and proteins that are specifically found in rod and cone photoreceptors and that may be differentially expressed during age-related and juvenile macular degeneration. Finally, together with collaborators in the U.S. and Germany, we are exploring novel gene transfer approaches to rescue animal models of Stargardt’s macular degeneration and X-linked juvenile retinoschisis from loss in vision.
We are investigating the role of this protein in outer segment structure and morphogenesis. RS1 or retinoschisin is a discoidin containing extracellular protein implicated in retinal cell adhesion and associated with X-linked juvenile retinoschisis (Fig 9). We are studying the structure of this protein and how mutations affect its folding pattern and subunit assembly. As part of our research program, we are also carrying out genomic and proteomic studies to identify genes and proteins that are specifically found in rod and cone photoreceptors and that may be differentially expressed during age-related and juvenile macular degeneration. Finally, together with collaborators in the U.S. and Germany, we are exploring novel gene transfer approaches to rescue animal models of Stargardt’s macular degeneration and X-linked juvenile retinoschisis from loss in vision.


